1.0 Introduction
Generic pharmaceutical products are clone of innovator products in the term of actives, strengths, dosage forms, dosage regimens and administrations. Actual fact is that innovator companies are very less in numbers in the world due to requirement of huge investment. Innovator companies are also not in good numbers enough to serve their medicines to all the worldwide patients. Reasonably, generic companies are established to serve their local patients with medicines at a manageable price. Generic companies cannot change strengths, dosage forms, dosage regimens, administration of a drug substance which are already researched and proved safe & effective via clinical trial by Innovator Company, rather generic company can play with excipients and physical attributes (appearance, shape, size, color, etc) of dosage form only.
2.0 Generic Products VS Innovator Products/Reference product
Innovator products have been approved as a new drug, or a drug that corresponds to one. Innovator products itself are considered as a reference product, besides reference product for Bio-equivalence study may be from other brand companies. Actually other brand product which BE study carried out with respect to innovator product will act as reference product. Innovator products itself is a reference product. Generic Products are the products of which active ingredients, strengths, dosage forms, and dosage regimens are the same as those of innovator products. Generic Products and Innovator Products differ in drug particle size, excipients, manufacturing process, equipment, site of manufacturing, batch size, but generic product should be equivalent with Innovator product in term of bioavailability. Bioavailability means the rate and extent of absorption of active ingredients or active metabolites from a drug product into the systemic circulation. When bioavailability of generic product and reference product are almost similar for same group of people, that case generic product is bioequivalent to that reference product which is already recognized as a reference product in the regulatory authority. Bio-equivalent study of generic product, known as ‘test product’ will be carried out with respect to brand product, known as reference product. Reference product will be from innovator company or other brand company. Regulatory authority will decide which one will be considered as a ‘Reference product’. One can find list of reference product in Orangebook (http://www.accessdata.fda.gov/scripts/cder/ob/ default.cfm).
3.0 Bioequivalence: why required
Bioequivalence ensures a generic product how much it is as close as reference product in term of bioavailability. Excipients used as well as physical attributes (particle size, process, physical parameters such as hardness for tablet) are release rate limiting factor for active ingredient from dosageform. In order to be bioequivalent (BE) with reference product, generic product should have in-vitro release rate of active ingredient from its dosageform as much closely as reference product. International drug regulatory authority urges to conduct Bioequivalence for ensuring to get better therapeutic activity from generic drug products.
[wp_ad_camp_1]
4.0 Category of Active Ingredients which require BE Study
The Biopharmaceutics Classification System is a guide for predicting the intestinal drug absorption provided by the U.S. Food and Drug Administration. This system restricts the prediction using the parameters solubility and intestinal permeability. The solubility classification is based on a United States Pharmacopoeia (USP) aperture. The intestinal permeability classification is based on a comparison to the intravenous injection. All those factors are highly important because 85% of the most sold drugs in the United States and Europe are orally administered. According to the Biopharmaceutics Classification System, drug substances are classified as follows: Class I – high permeability, high solubility (Eample: Metoprolol, the compounds of Class I are well absorbed and their absorption rate is usually higher than excretion). Class II – high permeability, low solubility (Example: Glibenclamide, the bioavailability of those products is limited by their solvation rate. A correlation between the in vivo bioavailability and the in vitro solvation can be found). Class III – low permeability, high solubility (Example: Cimetidine, the absorption is limited by the permeation rate but the drug is solvated very fast. If the formulation does not change the permeability or gastro-intestinal duration time, then class I criteria can be applied). Class IV – low permeability, low solubility (Example: Hydrochlorothiazide, the compounds of Class IV have a poor bioavailability. Usually they are not well absorbed over the intestinal mucosa and a high variability is expected).
5.0 In-vitro Dissolution Profile In-vitro dissolution is a pre-assessment tool prior to conducting Bioequivalence study in order to see how much release of active drug of a generic product from its dosageform is as close as that of a reference product from its dosageform. This in-vitro dissolution will act as pre-assessment tool before conducting BE study. In-vitro dissolution profile is the % dissolution of active ingredient from its dosageform at sampling time interval in chemical laboratory. In-vitro dissolution profile is carried out for both generic product and reference product and their release is characterized by similarity factor (f2) and dissimilarity factor (f1).
5.1 Dissolution tests
Dissolution tests should be performed, using a suitably validated dissolution system and standard method.
- Number of vessels: 12 vessels or more under each testing condition.
- Testing conditions:The test should be carried out under the following conditions.
- Apparatus: Pharmacopeial paddle apparatus.
- Volume of test solution: Basically 900 ml.
- Temperature: 370C ± 0.50C
- Paddle speed : As per Pharmacopeia or Dissolution Methods of all drug products in FDA database.
- Dissolution Medium: As per Pharmacopeia or Dissolution Methods of all drug products in FDA database.
- Sampling Time Interval (Profile) : As per Pharmacopeia or Dissolution Methods of all drug products in FDA database.
If testing method is not available in Pharmacopeia or Dissolution Methods of all drug products in FDA database, that case the first and second media for the dissolution test are used as pH 1.2 and 6.8 test solutions, respectively. Diluted McIlvaine buffers (pH is adjusted by 0.05mol/L disodium hydrogen phosphate and 0.025mol/L citric acid) are used for other pH solutions. If the average dissolution rate of the reference product does not reach 85% by six hours under any of the above-mentioned dissolution test conditions but does reach 85% in another suitable dissolution media, a test using the other dissolution media may be added.
| A) Drug Products containing acidic active substance | ||
| Paddle speed (rpm) | Medium pH fordissolution test | Remarks |
| 50a)100C) | (1) 1.2(2) 5.5 – 6.5b)(3) 6.8 – 7.5b)(4) Water(1), (2), or (3)b) | a) The paddle method at 75 rpm or the basket method at 100 rpm can be usedinstead of the paddle method at 50 rpm, when coning phenomenon of disintegratesin the bottom of vessel is observed.b) In the dissolution media where the average dissolution of reference product reaches 85% within the testing time specified, the dissolution medium where the dissolution is the slowest should be selected. When the average dissolution of the reference product does not reach 85 % within the specified time in any of dissolution media, the dissolution medium should be selected where the dissolution is the fastest. c) If, in a dissolution test by the paddle method at 50 or 75 rpm, both the referenceproduct and the test product dissolve an average of 85% or more within 30 minutes in dissolution medium that could be used with the paddle method at 100 rpm, the dissolution test by the paddle method at 100 rpm may be omitted. |
| B) Drug Products containing neutral or basic active substance and coated products | ||
| Paddle speed (rpm) | Medium pH fordissolution test | Remarks |
| 50a)100C) | (1) 1.2(2) 3.0 – 5.0b)(3) 6.8b)(4) Water(1), (2), or (3)b) | a) The paddle method at 75 rpm or the basket method at 100 rpm can be usedinstead of the paddle method at 50 rpm, when coning phenomenon of disintegratesin the bottom of vessel is observed.b) In the dissolution media where the average dissolution of reference product reaches 85% within the testing time specified, the dissolution medium where the dissolution is the slowest should be selected. When the average dissolution of the reference product does not reach 85% within the specified time in any of dissolution media, the dissolution medium should be selected where the dissolution is the fastest. c)If, in a dissolution test by the paddle method at 50 or 75 rpm, both the referenceproduct and the test product dissolve an average of 85% or more within 30 minutes in dissolution media that could be used with the paddle method at 100 rpm, the dissolution test by the paddle method at 100 rpm may be omitted. |
| C) Drug Products containing poorly soluble active substanceA drug product containing a poorly soluble drug is a drug product for which, when the test is performed at 50 rpm, the average dissolution rate of the reference product does not reach 85% within the designated test time in any of the dissolution media specified in 1) or in 2) above with no surfactant in the medium. | |||
| Paddle speed (rpm) | Medium pH fordissolution test | Surfactant | Remarks |
| 50a)100C) | (1) 1.2(2) 4.0(3) 6.8(4) Water(5) 1.2(6) 4.0(7) 6.8(5), (6), or (7)d) | NonNonNonNonPolysorbate 80b)Polysorbate 80b)Polysorbate 80b)Polysorbate 8e) | a) The paddle method at 75 rpm or the basket method at 100 rpm can be used instead of the paddle method at 50 rpm, when coning phenomenon of disintegrates in the bottom of vessel is observed.b) Investigate polysorbate 80 concentrations of 0.01%, 0.1%, 0.5% and 1.0% (W/V). Determine the lowest polysorbate 80 concentration necessary for the reference product to dissolve an average of 85% or more in at least one of dissolution media (5), (6) and (7) within the designated test time, and add thatconcentration of polysorbate 80 to dissolution medium (5), (6) or (7). If the reference product does not dissolve an average of 85% in any of the dissolution media within the designated test time, choose the polysorbate 80 concentration at which dissolution is fastest. In the event that polysorbate 80 affects the dissolution behaviour of the drug by interacting with the drug or with excipients, or in another such event, it is permissible to replace potassium dihydrogen phosphate with sodium dihydrogen phosphate as the buffering agent and use sodium lauryl sulfate. However, if sodium lauryl sulfate is used, the solubility of the drug may not exceed the solubility maximum concentration specified for polysorbate 80.c)If, in a dissolution test by the paddle method at 50 or 75 rpm, both the reference product and the test product dissolve an average of 85% or more within 30 minutes in dissolution medium that could be used with the paddle method at 100rpm, the dissolution test by the paddle method at 100 rpm may be omitted.d) In the test solutions where the average dissolution of reference product reaches 85% within the testing time specified, the dissolution media where the dissolution is the slowest should be selected. When the average dissolution of the reference product does not reach 85 % within the specified time in any of dissolution media, the dissolution medium should be selected where the dissolution is the fastest.e)The same concentration as that in the case of 50 rpm. |
| D) Drug Products containing enteric coating | ||
| Paddle speed (rpm) | Medium pH fordissolution test | Remarks |
| 50a)100b) | (1) 1.2(2) 6.0(3) 6.8(2) | a) The paddle method at 75 rpm or the basket method at 100 rpm can be usedinstead of the paddle method at 50 rpm, when coning phenomenon of disintegratesin the bottom of vessel is observed.b) If, in a dissolution test by the paddle method at 50 or 75 rpm, both the referenceproduct and the test product dissolve an average of 85% or more within 30 minutesin dissolution medium that could be used with the paddle method at 100 rpm, thedissolution test by the paddle method at 100 rpm may be omitted.Note:Enteric-coated products containing poorly soluble drugs should be tested by addingpolysorbate 80 to the dissolution media (2) and (3) according to the dissolution test method for products containing poorly soluble drugs as described above. |
5.2. Acceptance criteria for Similarity of dissolution profiles
The average dissolution rate of the test product (Generic product) is compared with the average dissolution rate of the reference product. If dissolution of the reference product or test product has a lag time, the dissolution curve can be adjusted with the dissolution lag time (Appendix 2). Criteria (1) to (3) below are applied after the lag time. However, when dissolution curves are corrected, the difference between the average dissolution lag times of the test product and reference product must be not more than 10 minutes. The time points for comparing dissolution rates when assessment is performed by the f2 function are specified in Appendix 1. 2.
[wp_ad_camp_3]When any of the criteria below are met under all the sets of dissolution test conditions, the dissolution behaviour is judged as similar. However, the average dissolution rate of the reference product must reach 85% or more within the designated test time under at least one set of dissolution test conditions.
If the comparison time point is to be less than 15 minutes, dissolution behaviour may be evaluated using a comparison time point of 15 minutes. If correction for lag time is performed, the comparison time point of 15 minutes is the time before correction. If the pH of the dissolution test medium is 1.2 for an enteric-coated product, dissolution behaviour may be evaluated using only the dissolution rate at the designated test time (after 2 hours).
A judgement of similarity in dissolution does not mean bioequivalence.
(1) When the average dissolution of the reference product reaches 85% within 15 min: the average dissolution of the test product reaches 85% within 15 min or is within that of the reference product ± 15 % at 15 min.
(2) When the average dissolution of the reference product reaches 85% at between 15 and 30 min: the average dissolution of the test product are within that of the reference product ± 15 % at two appropriate time points when the average dissolution of the reference product are around 60% and 85%. Or f2 value is not less than 42.
(3) When the average dissolution of the reference product does not reach 85% within 30min: the results meet one of the following criteria.
a. When the average dissolution of the reference product reaches 85% within the testing time specified: the average dissolution of the test product are within that of the reference product ± 15 % at two appropriate time points when the average dissolution of the reference product are around 40% and 85%. Or f2 value is not less than 42.
b. When the average dissolution of the reference products reaches 50 % and does not reaches 85 % within the testing time specified: the average dissolution of the test product are within that of the reference product ± 12 % at the testing time specified and at an appropriate time point when the average dissolution of the reference product reaches about a half of the average dissolution at the testing time specified. Or f2 value is not less than 46.
c. When the average dissolution of the reference product does not reach 50% within the testing time specified: the average dissolution of the test product are within that of the reference product ± 9 % at the testing time specified and at an appropriate time point when the average dissolution of the reference product is about a half of the average dissolution at the testing time specified. Or f2 value is not less than 53. However, when the average dissolution of the reference product is not more than 10% at the stipulated dissolution time, the average dissolution of the test product is within that of the reference product ± 9 % at the testing time specified only.
5.3 Graphs of in-vitro dissolution profile
 |
 |


6.0 Conclusion
In-vitro dissolution profile of generic product and reference product is done in chemical laboratory prior to conducting Bio-equivalence study. Dissolution profile of generic product and reference product are characterized by similarity factor (f2) and dissimilarity factors (f1). Dissolution profile signifies how much similarity and dissimilarity are present between the % dissolved of active ingredient from its dosageform for both generic product and reference product.
7.0 References
- Guidance for Industry: Bioavailability and BioequivalenceStudies for Orally Administered DrugProducts — General Considerations; [Online] Available from: www.fda.gov/downloads/Drugs/…/Guidances/[Accessed 04 Feb 2013]
- English translation of Attachment 1 of Division-Notification 0229 No. 10 of the Pharmaceutical and Food Safety Bureau, dated February 29, 2012 ; [Online] Available from: www.nihs.go.jp/drug/be-guide(e)/Generic/ [Accessed 04 Feb 2013]
- Bioequivalence Study Design Considerations – Dr. John Gordon, Prequalification of Medicines Programme: 2nd Meeting with Medicine, Geneva April 5, 2011; [Online] Available from: http://apps.who.int/prequal/trainingresources/pq_pres/ stakeholders_2011/ presentations/Day_2/ [Accessed 09 Feb 2013]
- Biopharmaceutics Classification System – Wikipedia [Online] Available from: http://en.wikipedia.org/wiki/ Biopharmaceutics_Classification_System [Accessed 04 Feb 2013]
- Orange Book: Approved Drug Products with Therapeutic Equivalence Evaluations; [Online] Available from: http://www.accessdata.fda.gov/scripts/cder/ob/ default.cfm [Accessed 04 Feb 2013]
- Dissolution Methods of all drug products in FDA database. [Online] Available from: http://www.fda.gov/Drugs/InformationOnDrugs /ucm135742.htm [Accessed 04 Feb 2013]
8.0 Appendix
Appendix 1. f2 (similarity factor) and time points for comparisons
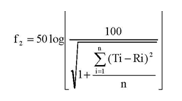
(1) Definition of f2
The following equation defines f2. Ti and Ri show the average dissolutions of the test and reference products at the time point (i), respectively, and n is the number of time points at which the average dissolution are compared.

(2) Time points for f2
1) When the average dissolution of the reference product reaches 85% (80% for extended release products)between 15 and 30 min: 15, 30, 45 min.
2) When the average dissolution of the reference product reaches 85% (80% for extended release products) between 30 min and the testing time point specified: at Ta/4, 2Ta/4, 3Ta/4 and Ta, where Ta is the time point at which average dissolution of the reference product reaches approximately 85% (80% for extended release products).
3) When the average dissolution of the reference product does not reach 85% (80% for extended release products) within the testing time point specified: at Ta/4, 2Ta/4, 3Ta/4 and Ta, where Ta is the time point at which average dissolution of the reference product reaches approximately 85% (80% for extended release products) of the amount dissolved at the testing time point specified.
Appendix 2. Adjusting dissolution curves with lag times
The lag time is defined as the time when 5% of the labeled claim of the active ingredient dissolves from the product. A lag time should be determined for respective product by linear interpolation, and then respective dissolution curve is obtained by adjusting dissolution curve with the lag time. Average dissolution curves of the test and reference products are obtained, which can be used for the assessment of similarity and equivalence in dissolution curves.
[wp_ad_camp_1]
About the Author:
Mohammad Dulal, Industrial Pharmacist licensed in MOH/Dubai, Senior Scientist, Product Development Lab, Julphar Gulf Pharmaceutical Ltd, Ras Al Khaimah, United Arab Emirates.
Cite this Article as:
Dulal, M. (2013) In-vitro Dissolution Profile acts as a pre-assessment tool for Bio-equivalence Study of Generic Pharmaceutical Oral Solid Products. [Online] Available from: http://pharmamirror.com/pharmaceutical-articles/bio-equivalence-study-of-generic-pharmaceutical-oral-solid-products/ [Accessed 19th Feb 2013]