Chinese pharmaceutical market is growing significantly and is going to be the world’s largest market within very few years. Many world renowned pharmaceutical companies have been expanding their footprint in efforts to enter the Chinese drug market as the government has adopted new policy to promote and favor drugs innovation.
The China Food and Drug Administration (CFDA) which was known as The State Food and Drug Administration (SFDA) until 2013 is the agency responsible for controlling this emerging pharma market. Their main responsibilities include drug approvals before introducing any pharmaceutical products and medical devices in China and post-marketing surveillance according to rules and laws to assure drug safety.
Seven Major affiliates of CFDA
- National Institute for the control of Pharmaceuticals and Biological Products: testing of drugs and medical devices, etc. as the top technical arbitration institute for drug quality in China
- Center for Drug Evaluation of SFDA: technical evaluation for drug registration
- Center for Drug Certification of SFDA: on-site inspection for GAP, GCP, GLP, GMP certification
- National Committee on the Assessment of the Protected Traditional Chinese Medical Products (Center for Health Food Evaluation of SFDA): technical assessment of the protected traditional Chinese medical products ; technical evaluation for market authorization of health food
- Center for Drug Reevaluation of SFDA: post-marketing surveillance through adverse reaction monitoring, reevaluation and elimination of drugs
- Center for Medical Device Evaluation of SFDA: technical evaluation for medical devices registration
- Certification Center for Licensed Pharmacist of SFDA: Licensed pharmacist registration, further education, etc.
Clinical Trials
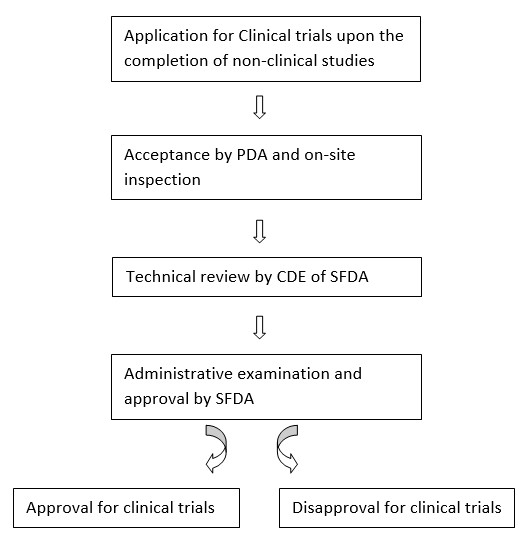
For a new drug approval or for a generic drug registration in China, clinical trial shall be conducted according to the requirements in the Annex of the Provisions. Any clinical trial application including bioequivalence study must be approved by CFDA and conducted in compliance with the Good Clinical Practice (GCP).
Upon completion of non-clinical studies, a clinical trial application could be sent to CFDA and on-site inspection is conducted following a technical review of CDE of CFDA. Then CFDA takes decision about the approval of conducting clinical trial.
According to SFDA Order no. 3, specified in Chapter 3: Article 5; sufficient scientific data and ethics must be established to conduct clinical trial method and the benefits must overweigh the risk of human subjects and public health.

Expedite Adverse Event Reporting
The protocol of the trial must be approved by Ethics committee and any serious adverse events that occur during the trial shall be reported to the Ethics Committee in time. The investigator shall ensure that the human subjects receive proper treatment when adverse event occurs during the trial. [SFDA order No. 3: Provisions for Drug Registration: GCP, Chapter 5, Article: 24-25]
If any serious adverse event occurs during the clinical trial, the investigators shall report to the drug regulatory departments of the relevant provinces, autonomous regions or municipalities directly under the Central Government and the China Food and Drug Administration and notify the applicant within 24 hours, and report to the ethic committee in time. [SFDA Order No. 28, Chapter III, Article: 41]
If there is any large-scale of unexpected adverse reaction or serious adverse event, the CFDA may order to change the protocol or to suspend or terminate the clinical trial. It is also the same for a serious adverse event that is not reported within the specified timeline. [SFDA Order No. 28, Chapter III, Article: 42-43]
When conducting an international multi-center clinical trial in China, if there are any observed serious adverse reaction and unexpected adverse reaction associated with the drug in any country, the applicant shall, in accordance with relevant regulations, report to the China Food and Drug Administration in time; [SFDA Order No. 28, Chapter III, Article: 44]
Periodic Safety Reporting
- The China Food and Drug Administration set an observation period of five years for new drugs to ensure drug safety.
- A drug manufacturer shall submit a report of thorough investigate of the manufacturing processes, quality, stability, therapeutic effect and adverse reactions, etc. annually.
- During drug manufacturing, distribution, use, testing or supervision, if any critical quality problem, serious or unexpected adverse reaction of a new drug is found, they shall report to the respective drug regulatory department and the department shall report to the China Food and Drug Administration after prompt investigation.
[SFDA Order No. 28, Chapter IV: Section 3, Article: 66-68]
Post Marketing
Expedite adverse event reporting
The China Food and Drug Administration shall be in charge of management of adverse drug reaction reporting and monitoring and The National Adverse Drug Reaction Monitoring Center shall be in charge of the technical work of the adverse drug reaction reporting and monitoring across the nation.
Chapter III: Section II: Individual Adverse Drug Reaction
- The drug manufacturer, distributor and medical institutions when become aware of an adverse drug reaction, they shall report an Adverse Drug Reaction form.
- Domestic and imported drugs within the safety period of five years must report all adverse reactions, in other cases only report new and serious events.
| ADR Type | Reporting Requirement | Recipient |
| New or serious drug adverse reaction | 15 working days | Local adverse drug reaction monitoring body |
| Death case | immediately | Provincial adverse drug reaction monitoring body |
| Other drug adverse reaction | 30 working days | Local adverse drug reaction monitoring body |
| It is required to timely report in the case of the follow-up information | ||
Chapter III, Section III: Group Adverse Drug Events
- The drug manufacturers, the drug distributors and the medical institutions shall, immediately upon awareness of any group adverse drug event, report to the county level drug regulatory department, health administrative department and adverse drug reaction monitoring body in their locality by telephone or fax. They shall fill and report few forms in for every case, and submitted through the adverse drug reaction monitoring information network.
- The drug manufacturers shall, upon awareness of any group adverse drug events, immediately investigate the group adverse drug event and shall prepare an investigation report within 7 days submit the same to the provincial food and drug administration department and adverse drug reaction monitoring body where it is
Chapter III, Section IV: Oversea Serious Adverse Drug Reactions
- In case of oversea serious adverse drug reactions relating to an imported drug or a domestic drug the drug manufacturers shall fill out the “Overseas Adverse Drug Reaction / Event Report Form”, and submit to the National Adverse Drug Reaction Monitoring Center within 30 days upon awareness. The drug manufacturers shall submit original reports and relevant information within 5 days as requested by National Adverse Drug Reaction Monitoring Center (NADMC).
- The NADMC shall make analysis and evaluation of the adverse drug reaction reports received by them, and report to the China Food and Drug Administration and the Ministry of Health every six months.
- In case an imported drug or a domestic drug is suspended from marketing or using or is withdrawn from an overseas market due to the adverse drug reaction, the drug manufacturers shall submit a written report to the China Food and Drug Administration and the National Adverse Drug Reaction Monitoring Center within 24 hours after their awareness.
Periodic Safety Reporting
The drug manufacturers shall make periodic review and analysis of the adverse reaction reports and monitoring data concerning their drugs. They shall summarize safety information collected from both domestic and abroad, perform risk and benefit assessment, and prepare periodic safety update reports.
The manufacturers of domestic drugs and imported drugs shall submit a periodic safety update report every year after they obtain the marketing license until the first re-registration date to the provincial adverse drug reaction monitoring body and National Adverse Drug Reaction Monitoring Center, respectively. Thereafter, they shall submit a periodic safety update report every 5 years.
For other domestic drugs, a periodic safety update report shall be submitted every 5 years. Each periodic safety update report shall be submitted within 60 days after expiration date of data collection.
The National Adverse Drug Reaction Monitoring Center shall summarize, analyze and evaluate the periodic safety update reports received by it, and submit the analysis and evaluation results of the previous year to the China Food and Drug Administration and the Ministry of Health on or before July 1 of each year.
[Administrative Regulation on Reporting and Monitoring of Adverse Drug Reaction (Directive of Ministry of Health No. 81, Chapter: 5, Article 36-40)
Further Reading:
China Good Clinical Practice, Available from: http://www.bioon.com/drug/chemdrug/243155.shtml. Accessed on: 5 April 2014.
Provisions for Drug Registration: (SFDA Order No. 28), Available from: http://eng.sfda.gov.cn/WS03/CL0768/61645.html , Accessed on: 4 April 2014.